Query String: AMPICILLIN
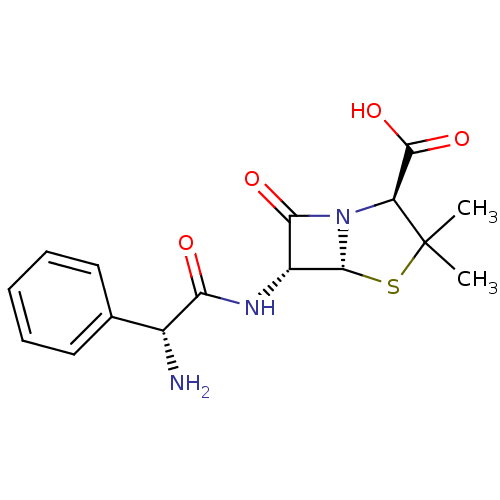 Aminobenzylpenicillin Ampicillin sulfate Amcill AMPICILLIN AY-6108 BDBM50350465
Aminobenzylpenicillin Ampicillin sulfate Amcill AMPICILLIN AY-6108 BDBM50350465
- ChEBML_40701 Inhibitory activity against Beta-lactamase type OXA1 (penicillinase) from Escherichia coli OXA1 using ampicillin (40 uM) as a substrate
- ChEBML_40865 Inhibitory activity against Beta-lactamase type OXA1 (penicillinase) from Escherichia coli OXA1 using ampicillin (40 uM) as a substrate
- ChEMBL_2234675 (CHEMBL5148447) Antibacterial activity against Enterococcus faecalis incubated for 16 to 24 hrs in presence of ampicillin by broth microdilution based spectrophotometric analysis
- Esterase Activity Assay CA activity was assayed according to method of Verpoorte et al. [Verpoorte et al., J. Biol. Chem., 242:4221-4229] described previously by Innocenti et al. [Innocenti et al., Bioorg. Med. Chem., 18:2159-2164; Innocenti et al., Bioorg. Med. Chem. Lett., 20:5050-5053] CA activity was determined by following thechange in absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25°C using a spectrophotometer (CHEBIOS UV-VIS). The enzymatic reaction, in a total volume of 3.0 mL, contained 1.4 mL 0.05 M Tris-SO4 buffer (pH 7.4), 1 mL, 3 mM 4-nitrophenylacetate, 0.5 mL H2O and 0.1 mL enzyme solution. A reference measurement was obtained by preparing the same cuvette without enzyme solution. The inhibitory effects of ampicillin sulfate, ceftriaxone, ceftizoxime, ranitidine were examined. All compounds were tested in triplicate at each concentration used. Different inhibitor concentrations were used. HCA-I enzyme activities were measured for ceftriaxone (0.003-0.033 mM), ceftizoxime (0.017-0.096 mM), ranitidine (0.026-0.053 mM) at cuvette concentrations and HCAII enzyme activities were measured for ampicillin sulfate (0.038-0.095 mM), ceftriaxone (0.003-0.021 mM), ceftizoxime (0.026-0.061 mM) and ranitidine (0.032-0.058 mM) at cuvette concentrations.
- FRET Activity Assay Expression and purification of the FXR-LBD: An overnight preculture of a transformed E. coli strain was diluted 1:20 in LB-Ampicillin medium and grown at 30� C. to an optical density of OD600=0.4-0.6. Gene expression was then induced by addition of 0.5 mM IPTG. Cells were incubated an additional 6 h at 30� C., 180 rpm. Cells were collected by centrifugation (7000�g, 7 min, rt). Per liter of original cell culture, cells were resuspended in 10 mL lysis buffer (50 mM Glucose, 50 mM Tris pH 7.9, 1 mM EDTA and 4 mg/mL lysozyme) and left on ice for 30 min. Cells were then subjected to sonication and cell debris removed via centrifugation (22000�g, 30 min, 4� C.). Per 10 mL of supernatant 0.5 mL prewashed Glutathione 4B sepharose slurry (Qiagen) was added and the suspension kept slowly rotating for 1 h at 4� C. Glutathione 4B sepharose beads were pelleted by centrifugation (2000�g, 15 sec, 4� C.) and washed twice in wash buffer (25 mM Tris, 50 mM KCl, 4 mM MgCl2 and 1M NaCl). The pellet was resuspended in 3 mL elution buffer per liter of original culture (elution buffer: 20 mM Tris, 60 mM KCl, 5 mM MgCl2 and 80 mM glutathione added immediately prior to use as powder). The suspension was left rotating for 15 min at 4� C., the beads pelleted and eluted again with half the volume of elution buffer than the first time. The eluates were pooled and dialysed overnight in 20 mM Hepes buffer (pH 7.5) containing 60 mM KCl, 5 mM MgCl2 as well as 1 mM dithiothreitol and 10% (v/v) glycerol. The protein was analysed by SDS-Page.
- Inhibition of Protease Activity Assay Full-length cDNA of human MALT1 gene (GenBank accession No: AB026118.1) amplified by PCR was inserted in flame to a SalI site located downstream of GST gene in a pGEX6P3 vector (GE Healthcare Japan Corp.) to prepare a vector (hereinafter, referred to as a pGEX6P3-MALT1 vector). Subsequently, E. coli for protein expression (BL21-RIL-codon plus-DE3, Agilent Technologies, Inc.) was transformed with the pGEX6P3-MALT1 vector and then analyzed by ampicillin resistance screening and colony PCR to obtain an E. coli strain expressing recombinant GST fusion MALT1. Protein expression was induced with isopropyl-(3-thiogalactopyranoside. After the expression induction, E. coli precipitates were recovered by centrifugation from the E. coli culture solution, and the E. coli precipitates were homogenized and then centrifuged to obtain a supernatant. The supernatant was purified using GSTrap FF column (GE Healthcare Japan Corp.) to obtain recombinant GST fusion MALT1.B) Evaluation of Inhibition of Protease Activity of MALT1:To 89 μL of an enzyme solution (4.8 g/mL GST fusion MALT1, 50 mmol/L MES, 150 mmol/L NaCl, 10% sucrose, 0.1% CHAPS, 10 mmol/L dithiothreitol, and 1 mol/L tri-ammonium citrate) per specimen, 1 μL of a test compound (DMSO-diluted solution) of each concentration was added to prepare a mixed solution. The mixed solution was incubated at room temperature for 30 minutes, followed by the measurement of the fluorescence value of the mixed solution (fluorescence value of the first measurement) (Ex: 380 nm, Em: 460 nm; Envision (Perkin Elmer Inc.)). Next, 10 μL of 200 μmol/L substrate (Ac-LRSR-AMC, SM Biochemicals LLC) was added (final concentration: 20 μmol/L) to the mixed solution, and the mixture was reacted by incubation at 30� C. for 80 minutes, followed by the measurement of the fluorescence value of the reaction solution (fluorescence value of the second measurement) (Ex: 380 nm, Em: 460 nm; Envision (Perkin Elmer Inc.)).
- beta-lactamase assay Expression and Purification of Beta-Lactamases. For his tag KPC-2 beta-lactamase, bacteria were grown overnight at 30 C with shaking in 50 mL LB broth supplemented with 50 μg/mL kanamycin. Two liters of LB broth supplemented with 50 μg/mL kanamycin, 200 mM sorbitol, and 5 mM betaine were each inoculated with 10 mL of overnight bacterial culture. Cultures were then grown at 37 C until an optical density at 600 nm (OD600) of 0.6-0.7. Protein expression was then initiated by the addition of IPTG (final concentration 0.5 mM), followed by growth for 16 hr at 20 C. Cells were pelleted by centrifugation and stored at −80 C until further use. The his tag KPC-2 beta-lactamase was purified by nickel affinity chromatography and gel filtration. Briefly, the cell pellets were thawed and re-suspended in 40 mL of buffer A (20 mM Tris-HCl pH 8.0, 300 mM NaCl, 20 mM imidazole) with one complete protease inhibitor cocktail tablet (Roche) and disrupted by sonication, followed by ultracentrifugation to clarify the lysate. After ultracentrifugation, the supernatant was passed through a 0.22 μm filter before loading onto a 5 mL HisTrap HP affinity column (GE Healthcare Life Sciences, USA) pre-equilibrated with buffer A. His tag KPC-2 was eluted by a linear imidazole gradient (20 mM to 500 mM). Fractions were analyzed by SDS-PAGE. Fractions containing his tag KPC-2 were concentrated using a 10 k NMWL Amicon Ultra-15 Centrifugal Filter Unit. Concentrated his tag KPC-2 was then loaded onto a superdex 75 gel filtration column (GE Healthcare Life Sciences) pre-equilibrated with 20 mM Tris-HCl pH 8.0, 300 mM NaCl. Protein concentration was determined by absorbance at 280 using an extinction coefficient of 39,545. SDS-PAGE analysis indicated that the eluted protein was more than 95% pure.For sumo tag NDM-1 metallo-beta-lactamase, bacteria were grown overnight at 30 C with shaking in 50 mL LB broth supplemented with 100 μg/mL ampicillin. Two liters of LB broth supplemented with 100 μg/mL ampicillin were each inoculated with 10 mL of overnight bacterial culture. Cultures were then grown at 37 C until an optical density at 600 nm (OD600) of 0.6-0.7. Protein expression was then initiated by the addition of IPTG (final concentration 0.5 mM), followed by growth for 16 hr at 20 C. Cells were pelleted by centrifugation and stored at −80 C until further use. The sumo tag NDM-1 beta-lactamase was purified by nickel affinity chromatography and gel filtration. Briefly, the cell pellets were thawed and re-suspended in 40 mL of buffer A (20 mM HEPES pH 7.4, 0.5 M NaCl, 20 mM imidazole) with one complete protease inhibitor cocktail tablet (Roche) and disrupted by sonication, followed by ultracentrifugation to clarify the lysate. After ultracentrifugation, the supernatant was passed through a 0.22 μm filter before loading onto a 5 mL HisTrap HP affinity column (GE Healthcare Life Sciences, USA) pre-equilibrated with buffer A. Sumo tag NDM-1 was eluted by a linear imidazole gradient (20 mM to 500 mM). Fractions were analyzed by SDS-PAGE. Fractions containing sumo tag NDM-1 were buffer exchanged into 20 mM HEPES pH 7.0, 100 mM NaCl. Cleavage of the sumo tag was then carried out with ULP1 protease overnight at room temperature and then concentrated using a 10 k NMWL Amicon Ultra-15 Centrifugal Filter Unit. The sample was then loaded back onto a nickel affinity column and the flow through was collected, containing the untag NDM-1. NDM-1 was concentrated and loaded onto a gel filtration column (GE Healthcare Life Sciences) pre-equilibrated with 20 mM HEPES pH 7.0, 100 mM NaCl. Protein concentration was determined by absorbance at 280 using an extinction coefficient of 27,960. SDS-PAGE analysis indicated that the eluted protein was more than 95% pure.Steady-State Kinetic Analysis. Steady-state kinetic parameters were determined by using a Biotek Cytation Multi-Mode Reader. For KPC-2, each assay was performed in 100 mM Tris-HCl pH 7.0,
- FRET Activity Assay The human FXRalpha LBD was expressed in E. coli strain BL21(DE3) as an N-terminally GST tagged fusion protein. The DNA encoding the FXR ligand binding domain was cloned into vector pDEST15 (Invitrogen). Expression was under control of an IPTG inducible T7 promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq). Expression and purification of the FXR-LBD: An overnight preculture of a transformed E. coli strain was diluted 1:20 in LB-Ampicillin medium and grown at 30� C. to an optical density of OD600=0.4-0.6. Gene expression was then induced by addition of 0.5 mM IPTG. Cells were incubated an additional 6 h at 30� C., 180 rpm. Cells were collected by centrifugation (7000�g, 7 min, rt). Per liter of original cell culture, cells were resuspended in 10 mL lysis buffer (50 mM Glucose, 50 mM Tris pH 7.9, 1 mM EDTA and 4 mg/mL lysozyme) and left on ice for 30 min. Cells were then subjected to sonication and cell debris removed via centrifugation (22000�g, 30 min, 4� C.). Per 10 mL of supernatant 0.5 mL prewashed Glutathione 4B sepharose slurry (Qiagen) was added and the suspension kept slowly rotating for 1 h at 4� C. Glutathione 4B sepharose beads were pelleted by centrifugation (2000� g, 15 sec, 4� C.) and washed twice in wash buffer (25 mM Tris, 50 mM KCl, 4 mM MgCl2 and IM NaCl). The pellet was resuspended in 3 mL elution buffer per liter of original culture (elution buffer: 20 mM Tris, 60 mM KCl, 5 mM MgCl2 and 80 mM glutathione added immediately prior to use as powder). The suspension was left rotating for 15 min at 4� C., the beads pelleted and eluted again with half the volume of elution buffer than the first time. The eluates were pooled and dialysed overnight in 20 mM Hepes buffer (pH 7.5) containing 60 mM KCl, 5 mM MgCl2 as well as 1 mM dithiothreitol and 10% (v/v) glycerol. The protein was analysed by SDS-Page.
- Inhibition LYP ActivityAssay Expression and Purification of the LYP Catalytic Domain N-terminal (His)6-tagged LYP catalytic domain (residues 1-303) was subcloned into pET28a. For protein expression, the LYP expressing construct was transformed into Escherichia coli BL21-(DE3). Transformed cells were grown at 37� C. in Luria broth (LB) containing 100 μg/mL ampicillin for 4 h until the OD600 reached 0.6 and then induced for growth overnight at room temperature with 0.4 mM IPTG. Cells were harvested by centrifugation (6000 rpm for 15 min at 4� C.), and the cell pellets from 1.5 L of LB medium were suspended in 30 mL of ice-cold lysis buffer consisting of 5 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.9), 0.05 mg/mL trypsin inhibitor, and 0.1 mM PMSF. The suspensions were passed twice through a French press at 1000 psi, and the cell lysates were centrifuged at 4� C. for 30 min at 15000 rpm. The supernatants were mixed with 2 mL of Ni-NTA agarose (His*Bind Resin) (Qiagen) at 4� C. for 1 h, and then the mixture was transferred to an empty column. The column was washed with 200 mL of binding buffer (5 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.9)), followed by 20 mL of wash buffer (20 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.9)), and then eluted with 20 mL of elution buffer (200 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl (pH 7.9), 5 mM DTT). The elution was dialyzed for 6 h at 4� C. against 1 L buffer A (50 mM NaCl, 20 mM MES (pH 5.8), 1 mM EDTA) and then loaded onto a Mono S column equilibrated at 4� C. with buffer A. The column was washed with 10 mL of buffer A and then eluted with a 40 mL of linear gradient of 0-1 M NaCl in buffer A. The column fractions were analyzed by measuring the absorbance at 280 nm and by carrying out SDS-PAGE analysis. The fractions were combined, concentrated at 4� C. to <1 mL using an Amicon concentrator, and then loaded onto a gel filtration column Superdex 75. The column was eluted with buffer A, and then the fractions which contained protein were combined and concentrated to 8 mg/mL and stored at −80� C. The LYP preparation was shown to be homogeneous by SDS-PAGE analysis.
- FRET Activity Assay Determination of a ligand mediated cofactor peptide interaction to quantify ligand binding to the nuclear receptor FXR was performed as follows.Preparation of human FXR alpha ligand binding domain: The human FXRalpha LBD was expressed in E. coli strain BL21(DE3) as an N-terminally GST tagged fusion protein. The DNA encoding the FXR ligand binding domain was cloned into vector pDEST15 (Invitrogen). Expression was under control of an IPTG inducible T7 promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq). Expression and purification of the FXR-LBD: An overnight preculture of a transformed E. coli strain was diluted 1:20 in LB-Ampicillin medium and grown at 30� C. to an optical density of OD600=0.4-0.6. Gene expression was then induced by addition of 0.5 mM IPTG. Cells were incubated an additional 6 h at 30� C., 180 rpm. Cells were collected by centrifugation (7000�g, 7 min, rt). Per liter of original cell culture, cells were resuspended in 10 mL lysis buffer (50 mM Glucose, 50 mM Tris pH 7.9, 1 mM EDTA and 4 mg/mL lysozyme) and left on ice for 30 min. Cells were then subjected to sonication and cell debris removed via centrifugation (22000�g, 30 min, 4� C.). Per 10 mL of supernatant 0.5 mL prewashed Glutathione 4B sepharose slurry (Qiagen) was added and the suspension kept slowly rotating for 1 h at 4� C. Glutathione 4B sepharose beads were pelleted by centrifugation (2000�g, 15 sec, 4� C.) and washed twice in wash buffer (25 mM Tris, 50 mM KCl, 4 mM MgCl2 and 1M NaCl). The pellet was resuspended in 3 mL elution buffer per liter of original culture (elution buffer: 20 mM Tris, 60 mM KCl, 5 mM MgCl2 and 80 mM glutathione added immediately prior to use as powder). The suspension was left rotating for 15 min at 4� C., the beads pelleted and eluted again with half the volume of elution buffer than the first time. The eluates were pooled and dialysed overnight in 20 mM Hepes buffer (pH 7.5) containing 60 mM KCl, 5 mM MgCl2 as well as 1 mM dithiothreitol and 10% (v/v) glycerol. The protein was analysed by SDS-Page.The method measures the ability of putative ligands to modulate the interaction between the purified bacterial expressed FXR ligand binding domain (LBD) and a synthetic biotinylated peptide based on residues 676-700 of SRC-1 (LCD2, 676-700). The sequence of the peptide used was B-CPSSHSSLTERHKILHRLLQEGSPS-COOH (SEQ ID NO: 1) where the N-terminus was biotinylated (B). The ligand binding domain (LBD) of FXR was expressed as fusion protein with GST in BL-21 cells using the vector pDEST15. Cells were lysed by sonication, and the fusion proteins purified over glutathione sepharose (Pharmacia) according to the manufacturers instructions. For screening of compounds for their influence on the FXR-peptide interaction, the Perkin Elmer LANCE technology was applied. This method relies on the binding dependent energy transfer from a donor to an acceptor fluorophor attached to the binding partner of interest. For ease of handling and reduction of background from compound fluorescence LANCE technology makes use of generic fluorophore labels and time resolved detection Assays were done in a final volume of 25 μL in a 384 well plate, in a Tris-based buffer (20 mM Tris-HCl pH 7.5; 60 mM KCl, 5 mM MgCl2; 35 ng/μL BSA), containing 20-60 ng/well recombinantly expressed FXR-LBD fused to GST, 200-600 nM N-terminally biotinylated peptide, representing SRC1 aminoacids 676-700, 200 ng/well Streptavidin-xlAPC conjugate (Prozyme) and 6-10 ng/well Eu W1024-antiGST (Perkin Elmer). DMSO content of the samples was kept at 1%. After generation of the assay mix and diluting the potentially FXR modulating ligands, the assay was equilibrated for 1 h in the dark at rt in FIA-plates black 384 well (Greiner). The LANCE signal was detected by a Perkin Elmer VICTOR2VTM Multilabel Counter.
- RET Activity Assay Preparation of human FXR alpha ligand binding domain: The human FXRalpha LBD was expressed in E. coli strain BL21(DE3) as an N-terminally GST tagged fusion protein. The DNA encoding the FXR ligand binding domain was cloned into vector pDEST15 (Invitrogen). Expression was under control of an IPTG inducible T7 promoter. The amino acid boundaries of the ligand binding domain were amino acids 187-472 of Database entry NM_005123 (RefSeq). Expression and purification of the FXR-LBD: An overnight preculture of a transformed E. coli strain was diluted 1:20 in LB-Ampicillin medium and grown at 30� C. to an optical density of OD600=0.4-0.6. Gene expression was then induced by addition of 0.5 mM IPTG. Cells were incubated an additional 6 h at 30� C., 180 rpm. Cells were collected by centrifugation (7000�g, 7 min, rt). Per liter of original cell culture, cells were resuspended in 10 mL lysis buffer (50 mM Glucose, 50 mM Tris pH 7.9, 1 mM EDTA and 4 mg/mL lysozyme) and left on ice for 30 min. Cells were then subjected to sonication and cell debris removed via centrifugation (22000�g, 30 min, 4� C.). Per 10 mL of supernatant 0.5 mL prewashed Glutathione 4B sepharose slurry (Qiagen) was added and the suspension kept slowly rotating for 1 h at 4� C. Glutathione 4B sepharose beads were pelleted by centrifugation (2000�g, 15 sec, 4� C.) and washed twice in wash buffer (25 mM Tris, 50 mM KCl, 4 mM MgCl2 and 1M NaCl). The pellet was resuspended in 3 mL elution buffer per liter of original culture (elution buffer: 20 mM Tris, 60 mM KCl, 5 mM MgCl2 and 80 mM glutathione added immediately prior to use as powder). The suspension was left rotating for 15 min at 4� C., the beads pelleted and eluted again with half the volume of elution buffer than the first time. The eluates were pooled and dialysed overnight in 20 mM Hepes buffer (pH 7.5) containing 60 mM KCl, 5 mM MgCl2 as well as 1 mM dithiothreitol and 10% (v/v) glycerol. The protein was analysed by SDS-Page.The method measures the ability of putative ligands to modulate the interaction between the purified bacterial expressed FXR ligand binding domain (LBD) and a synthetic biotinylated peptide based on residues 676-700 of SRC-1 (LCD2, 676-700). The sequence of the peptide used was B-CPSSHSSLTERHKILHRLLQEGSPS-COOH (SEQ ID NO: 1) where the N-terminus was biotinylated (B). The ligand binding domain (LBD) of FXR was expressed as fusion protein with GST in BL-21 cells using the vector pDEST15. Cells were lysed by sonication, and the fusion proteins purified over glutathione sepharose (Pharmacia) according to the manufacturers instructions. For screening of compounds for their influence on the FXR-peptide interaction, the Perkin Elmer LANCE technology was applied. This method relies on the binding dependent energy transfer from a donor to an acceptor fluorophor attached to the binding partner of interest. For ease of handling and reduction of background from compound fluorescence LANCE technology makes use of generic fluorophore labels and time resolved detection Assays were done in a final volume of 25 μL in a 384 well plate, in a Tris-based buffer (20 mM Tris-HCl pH 7.5; 60 mM KCl, 5 mM MgCl2; 35 ng/μL BSA), containing 20-60 ng/well recombinantly expressed FXR-LBD fused to GST, 200-600 nM N-terminally biotinylated peptide, representing SRC1 aminoacids 676-700, 200 ng/well Streptavidin-xlAPC conjugate (Prozyme) and 6-10 ng/well Eu W1024-antiGST (Perkin Elmer). DMSO content of the samples was kept at 1%. After generation of the assay mix and diluting the potentially FXR modulating ligands, the assay was equilibrated for 1 h in the dark at rt in FIA-plates black 384 well (Greiner). The LANCE signal was detected by a Perkin Elmer VICTOR2V Multilabel Counter. The results were visualized by plotting the ratio between the emitted light at 665 and 615 nm. A basal level of FXR-peptide formation is observed in the absence of added ligand.